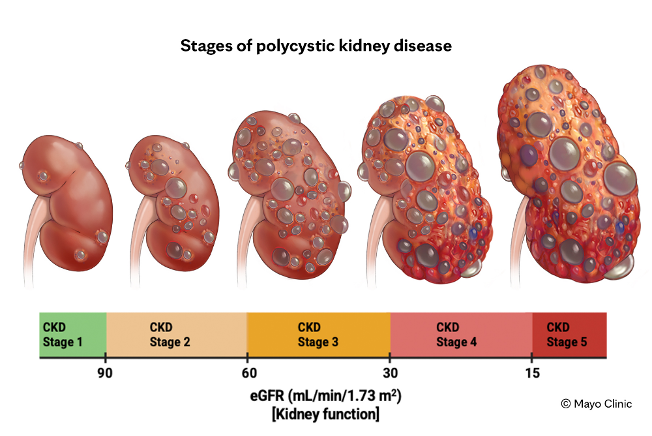
Polycystic kidney disease (PKD) is widely understood as a progressive genetic disorder characterised by the relentless formation and expansion of fluid-filled cysts within the kidneys. Over time, these cysts multiply and enlarge, gradually displacing healthy renal tissue and driving progressive loss of kidney function. PKD can also affect the liver and pancreas, and is associated with increased rates of hypertension, kidney stones, and end-stage renal disease. Patients with PKD also carry an elevated risk of various cancers, a relationship that, until recently, was poorly understood.
But beneath this conventional framing lies a far more intriguing biology. Emerging research and a growing body of clinical observation suggests that cyst formation in PKD shares deep mechanistic similarities with cancer cell metabolism and that both conditions may be driven, at least in part, by the same upstream trigger: a disrupted gut microbiome, bacterial endotoxin burden, and fungal overgrowth activating a fundamental cellular defence mechanism known as the Cell Danger Response (CDR).
Cancer is not solely a disease of uncontrolled cell division; it is also a disease of profoundly altered metabolism. Among the most important and well-studied of these metabolic changes is the Warburg effect, first described by German biochemist Otto Warburg in the 1920s.
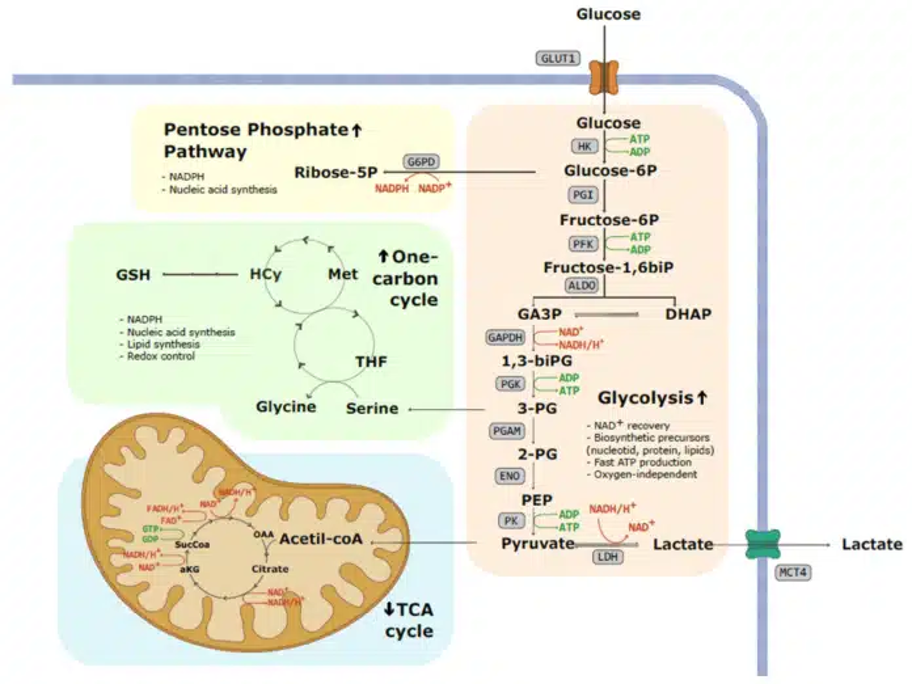
Under normal conditions, healthy cells produce energy through oxidative phosphorylation in the mitochondria, an oxygen-dependent, highly efficient process that yields approximately 32–36 ATP molecules per glucose molecule. Cancer cells, however, largely abandon this pathway in favour of aerobic glycolysis: converting glucose to lactate even when oxygen is readily available. This process yields only 2 ATP molecules per glucose, an approximately 18-fold reduction in energy efficiency.
The critical biological consequence of this metabolic switch is that the body couples energy production with cellular growth. Because only 2 units of energy are produced instead of 36, the glycolytic process must be upregulated 18-fold to compensate, and this 18-fold increase in metabolic activity drives an 18-fold increase in cellular proliferation. This is the engine behind tumour growth.
Professor Thomas Seyfried’s landmark work (Cancer as a Metabolic Disease) established this metabolic framework for cancer. Subsequent researchers applied it to PKD around 2015, and the parallels are striking.
This shared metabolic dependency has a direct therapeutic implication: if cyst cells, like cancer cells, are dependent on glucose for their aberrant proliferative activity, then dietary interventions that reduce circulating glucose availability are a rational therapeutic strategy. This is the metabolic rationale underpinning the use of ketogenic and very-low-carbohydrate diets in both PKD management and integrative oncology, a point we return to below.
Image via Warburg Effect: A Key Metabolic Hallmark of Cancer Cells
Understanding Polycystins: The Proteins at the Root of PKD
To understand why PKD cysts form and grow, we must first understand the proteins whose dysfunction drives the process. The two key proteins Polycystin-1 (PC1) and Polycystin-2 (PC2) are encoded by the PKD1 and PKD2 genes, respectively, and together they constitute one of the most important regulatory systems in kidney tubule cell biology.
- Polycystin-1 (PC1): A large transmembrane protein expressed on the surface of kidney tubule cells and their cilia. PC1 functions as a mechanosensory receptor, detecting fluid flow through the tubules, and regulating downstream signalling pathways that govern cell growth, differentiation and apoptosis. In essence, PC1 instructs cells when to cease dividing.
- Polycystin-2 (PC2): A calcium-permeable ion channel that controls the entry of calcium ions into the cell. PC2 works in close coordination with PC1 and is essential for normal intracellular calcium homeostasis.
Together, PC1 and PC2 operate as a precisely coordinated signalling system. PC1 senses environmental cues; PC2 responds by modulating calcium entry. When either protein is deficient, as in heterozygous PKD1 or PKD2 mutations, intracellular calcium signalling is disrupted, cyclic AMP (cAMP) levels rise, and epithelial cells begin to proliferate and secrete fluid uncontrollably, forming and expanding cysts.
An important mechanistic nuance: in a heterozygous PKD mutation (one functional allele, one mutated allele), evidence suggests the mutated copy may frequently not be expressed, meaning the cell likely has approximately 50% of the normal polycystin level rather than producing a misfolded, pathological protein. This reframes the underlying problem as haploinsufficiency, a dosage issue that has different implications for therapeutic targeting than the production of an actively pathological protein would.
Critically, PC1 also functions as an epithelial adhesion regulator, contributing to the integrity of tight junctions, the cellular seals between adjacent epithelial cells that prevent molecules from leaking paracellularly. This tight junction function is directly relevant to the endotoxin-cyst trigger mechanism discussed in the following section.
One of the most compelling and clinically underappreciated lines of evidence connecting the gut microbiome to PKD concerns the role of bacterial endotoxins as initiating triggers for cyst growth.
What Are Endotoxins?
Endotoxins, also known as lipopolysaccharides (LPS), are structural components of the outer membrane of gram-negative bacteria. They are continuously shed in small quantities during normal bacterial metabolism and released in large amounts upon bacterial death. They are potent activators of the innate immune system, triggering robust inflammatory responses even at very low concentrations.
The Germ-Free Mouse Study: Microbiome as a Permissive Factor
A foundational 1980s study using a mouse model carrying the PKD-equivalent gene mutation demonstrated that when animals were raised in a completely sterile, germ-free environment devoid of all gut bacteria, 96% of the mice showed no cyst development, despite carrying the causative gene mutation. This single finding fundamentally reframes PKD: the genetic mutation alone is insufficient to produce the disease. Something derived from the microbiome is required to activate cyst formation.
Endotoxin Is the Missing Trigger
Subsequent in vitro experiments attempted to generate PKD cysts from cultured mutant kidney cells. Mechanical cellular injury alone, using damaging chemical agents such as NDGA, was insufficient to provoke cyst formation. Cyst growth only occurred consistently when endotoxin was added alongside the injury stimulus. Injury plus endotoxin produced cysts; injury alone did not.
Extending this into the clinical setting, elevated endotoxin levels have been detected in the urine of PKD patients, providing direct evidence that endotoxin exposure is present and ongoing in this population. Separately, microbiome sequencing studies have identified specific bacterial species consistently absent from the gut flora of PKD patients compared to healthy controls, raising the possibility that certain microbial communities normally suppress endotoxin-driven cyst initiation, and that their absence is integral to disease pathogenesis.
Recall that polycystins regulate epithelial tight junction integrity. In PKD, reduced polycystin expression means the tubular walls of the kidney are abnormally porous. Toxins that would ordinarily be filtered cleanly through the tubule lumen instead escape through loosened cell junctions into the surrounding interstitial tissue, a space from which they cannot be cleared through normal physiological mechanisms.
This model is powerfully supported by a remarkable experimental observation: when researchers surgically removed a PKD cyst, placed it in culture, aspirated the cyst fluid, and repeatedly flushed out the toxic contents, the cyst tissue reverted to normal, functional kidney tissue within 72 hours. The cyst architecture itself was never inherently pathological. It was a response to a pathological stimulus, and when that stimulus was removed, normal biology reasserted itself.
Further evidence: cyst fluid from a PKD patient, when applied to healthy, non-PKD kidney tissue in culture, induces cyst formation in that tissue in the absence of any PKD gene mutation. This implies that once the endotoxin or toxic burden in cyst fluid reaches a sufficient concentration, it can override even a normal genetic background, initiating cyst formation as a protective response.
How Endotoxins Amplify Cyst Growth: The Molecular Pathways
- TLR4 activation: Endotoxins bind Toll-like receptor 4 (TLR4) on tubular epithelial cells, activating NF-κB and MAPK signalling cascades, upregulating pro-inflammatory cytokine production, enhancing epithelial proliferation and fluid secretion, and promoting fibrosis, all of which drive cyst enlargement.
- cAMP elevation: Endotoxin-driven inflammation indirectly raises intracellular cAMP levels in tubular cells. In PKD, where polycystin-mediated calcium signalling is already compromised, elevated cAMP is one of the central drivers of fluid accumulation and cyst expansion.
- Oxidative stress: Endotoxin exposure triggers the generation of reactive oxygen species (ROS), causing mitochondrial damage and further dysregulating the energy-production pathways already impaired in PKD cyst cells.
- Interstitial inflammation: Macrophage infiltration and cytokine signalling in the renal interstitium create a sustained pro-inflammatory microenvironment that interacts with PKD gene mutations to accelerate renal injury and cyst enlargement.
The Cell Danger Response: When Cells Abandon Energy Production to Fight
The Cell Danger Response (CDR) is a fundamental, evolutionarily conserved cellular defence mechanism first described by researcher Robert Naviaux. It provides a unifying framework for understanding why cells in both PKD and cancer shift to aerobic glycolysis, and why that shift is not an error but a purposeful biological strategy.
When a cell detects an internal threat from infection, toxins, trauma, or any perceived danger, and the immune system has failed to resolve that threat at the tissue level, the cell activates the CDR as its last line of defence. The mechanism involves redirecting the oxygen that would normally fuel oxidative phosphorylation toward the production of free radicals instead, which are then deployed as antimicrobial weapons against the intracellular invader. Without that oxygen, efficient ATP production ceases, and the cell shifts to aerobic glycolysis, the Warburg metabolic pattern.
The CDR also drives rapid cellular proliferation: more cells mean more capacity to generate free radicals and mount a broader immune response. In this framework, the “tumour” of early cancer and, arguably, the cyst of PKD may represent the body assembling an immune army around a contained, unresolved invader, using uncontrolled growth as a mechanism to amplify its defensive capacity.
Under normal circumstances, the CDR resolves successfully: the invader is neutralised, oxygen returns to the mitochondria, and normal oxidative metabolism resumes. It has been estimated that this process occurs and self-resolves in healthy people many times throughout life. But when the underlying trigger persists because the causative organism or toxin is not cleared, the CDR becomes chronically activated. Cells remain locked in defensive mode, intercellular communication breaks down, and the sustained metabolic shift becomes the disease itself.
Candida, Fungal Overgrowth & Their Role in Cancer and PKD
The CDR framework raises an important question: what intracellular invader is so persistently triggering cellular defence in cancer and, potentially, in PKD? Emerging research, most comprehensively synthesised by author Mark Lintern in Cancer Resolution, proposes a provocative answer: fungal infection, and specifically Candida, may be the initiating trigger in a far larger proportion of cancer cases than previously recognised. A panel of oncologists reviewed Lintern’s theory and endorsed it as scientifically credible.
The Proposed Mechanism
Candida is a commensal organism present in virtually all humans; under competent immune surveillance, it remains in a dormant or non-invasive state. But when local immunity is suppressed through radiation, antibiotic disruption, chronic psychological stress, nutritional deficiency, or immune dysregulation, Candida can begin to invade epithelial cells. The invaded cell activates the CDR, deploying free radicals to combat the fungal intruder. Oxygen is diverted from energy production. Aerobic glycolysis is activated. The cell danger response locks on, and if it cannot resolve because the fungal burden is not cleared, the sustained Warburg metabolic state and uncontrolled proliferation manifest as cancer.
Why the Ketogenic Diet May Be Insufficient Alone
The ketogenic diet is widely used in both integrative oncology and PKD management, and its metabolic rationale is sound: by severely restricting carbohydrate intake, it reduces circulating glucose, denies aerobic glycolysis the substrate it requires to drive abnormal growth, and creates conditions under which healthy cells can thrive on ketones while glucose-dependent cells struggle. Multiple PKD patients have documented measurable cyst reduction on serial ultrasound using ketogenic protocols.
However, a critical limitation emerges when we consider the fungal hypothesis. Candida can survive and continue to proliferate in a ketogenic metabolic environment. The ketogenic diet suppresses CDR-driven aerobic glycolysis, which manifests as visible tumour growth or cyst expansion. It does not eliminate the fungal trigger sustaining the CDR. The disease process may continue subclinically while appearing to resolve.
This has potentially serious implications. A well-documented clinical case illustrates the risk: a patient treated her brain tumour with an exceptionally strict ketogenic protocol over several years, achieving complete resolution on imaging. On relaxing dietary restriction while remaining in mild ketosis, she suffered a catastrophic recurrence and died within three months. The interpretation aligned with Lintern’s framework: the fungal burden, unchecked throughout the dietary intervention because the ketogenic state suppressed only its expression and not its presence, was free to trigger explosive regrowth when glucose availability increased slightly.
A second illustrative case: a cancer patient who had not responded to ketogenic diet therapy for a year subsequently received antifungal treatment for an unrelated indication and their cancer then responded.
Relevance to PKD
Published microbiome analyses of PKD cyst fluid have identified both endotoxins and fungal products in cyst contents, suggesting that fungal organisms may contribute to the intracystic toxic burden the cyst attempts to contain. Whether fungi serve as a primary driver of PKD cyst formation, analogous to their proposed role in cancer, remains to be formally investigated, but the biological plausibility is substantial.
Clinically, Candida and mycotoxin burden are frequently elevated in patients with complex chronic conditions, including CKD, chronic fatigue syndrome, post-infectious syndromes, and autoimmune disease. In integrative practice, screening for IgG and IgA antibody responses to Candida commonly reveals immune reactivity in 50–70% of patients, the majority of whom are asymptomatic by conventional standards. Comprehensive clearance of Candida and fungal burden, confirmed through testing rather than assumed, is therefore considered a foundational step before other therapeutic interventions, as active fungal infection occupies immune resources and limits the effectiveness of all subsequent strategies.
The Gut-Kidney Axis: Microbiome, Endotoxin Load & the FMT Hypothesis
Population-level gene sequencing studies have uncovered a striking statistical discrepancy in PKD. Ultrasound-based prevalence data predicts approximately 10 in 10,000 people would have PKD. Yet sequencing studies, including work from the York PI laboratory, find the PKD gene mutation present in approximately 20 in 10,000, twice the expected prevalence. The most parsimonious explanation: a significant proportion of people who carry the causative PKD gene mutation never develop clinically significant cysts, because the permissive microbiome trigger is absent.
This inference is supported by individual clinical observations. There are cases of individuals confirmed or suspected to carry the PKD mutation who, despite advanced age, have only a single small cyst or none at all. In such cases, the protective factor most likely to distinguish them from those with progressive disease is gut microbiome composition. Specifically, the absence of endotoxin-producing Gram-negative bacterial species, or the presence of species that actively suppress endotoxin translocation, may prevent the gut-kidney axis from delivering the cyst-initiating trigger to renal tubule cells.
Early microbiome colonisation is a strong determinant of adult gut flora composition. Infants born vaginally acquire diverse maternal microbial populations during delivery; those born by caesarean section do not. Breastfed infants receive prebiotic oligosaccharides that selectively cultivate bifidobacterial species including Bifidobacterium longum, consistently associated with longevity and gut integrity in blue zone population studies. These factors may meaningfully influence whether the PKD gene mutation ultimately expresses as clinical disease.
Faecal Microbiota Transplantation: An Emerging Therapeutic Horizon
If the gut microbiome is a primary permissive factor in PKD necessary for cyst initiation, then faecal microbiota transplantation (FMT) from individuals with the PKD mutation who remain cyst-free represents a compelling therapeutic hypothesis. By introducing a “protective” microbiome phenotype into PKD patients with established cysts, it may be possible to remove the endotoxin trigger and allow the body’s own resolution mechanisms to reduce cyst burden over time.
The proposed research pathway to test this hypothesis:
- Identify individuals with confirmed PKD gene mutation, aged 50 or older, with minimal or no cysts on ultrasound, the “protective microbiome phenotype”
- Perform comprehensive microbiome sequencing, comparing these individuals to PKD patients with progressive cyst disease
- Identify protective microbial species or the absence of endotoxin-producing gram-negative species
- Design FMT protocols using microbiome-matched donors and test cyst growth inhibition in germ-free PKD mouse models
- Translate to human clinical trials with microbiome composition as the primary therapeutic target
This research direction is being explored in collaboration with the Friedman Laboratory at the University of Seattle, where a patient-centred PKD research centre is being established to evaluate patient-proposed study concepts. It represents arguably one of the most promising and under-resourced areas of PKD research currently underway.
Cysts as Protective Biology: Why Blocking Them May Not Be the Answer
This emerging framework has profound implications for how we evaluate existing and proposed pharmacological approaches to PKD. Several therapeutic strategies have focused on chemically suppressing cyst growth directly, including vasopressin receptor antagonists such as tolvaptan, which reduce cAMP-driven cyst fluid secretion.
If, however, cyst formation is the body’s purposeful adaptive response to an unresolved endotoxin or toxic burden that has entered the renal interstitium through leaky tubules, then blocking cyst formation without addressing the underlying cause removes the containment mechanism without removing the toxin. The biological consequences of this toxin dispersal into renal tissue, escalating interstitial inflammation, and loss of the encapsulation response, may be more harmful than the cysts themselves.
The 72-hour cyst reversal experiment described earlier supports this interpretation: remove the toxic stimulus, and the cyst architecture resolves spontaneously into functioning kidney tissue. The cyst is not the enemy. It is the body’s solution to an enemy it cannot otherwise expel.
Clinical Implications: A Practical Integrative Framework
Synthesising the evidence across these interlocking mechanisms, the following clinical priorities emerge for integrative practitioners working with PKD patients:
- Fungal and mycotoxin assessment first: Urine mycotoxin panels, serum Candida IgG/IgA titres, and comprehensive stool fungal analysis should be considered standard investigative steps in PKD workup. HLA-DR genotyping identifies poor mycotoxin clearers who warrant more assertive antifungal management. Candida eradication, confirmed through serial testing rather than assumed, should precede other therapeutic layers.
- Gut barrier integrity: Reduce endotoxin translocation across the gut-kidney axis by supporting tight junction integrity and reducing gram-negative bacterial load through targeted dietary strategies, prebiotics (within renal tolerance), and barrier-supportive nutrients including zinc carnosine, glutamine, and butyrate.
- Endotoxin burden reduction: Dietary elimination of processed foods, refined carbohydrates and seed oils reduces the substrate load for gram-negative bacterial overgrowth. Comprehensive microbiome testing (GI360, CDSA, NutriPATH) can identify specific dysbiosis patterns relevant to endotoxin production.
- Ketogenic or very-low-carbohydrate diet with nuance: A ketogenic dietary protocol remains a valuable tool for suppressing glucose-dependent cyst metabolism and supporting overall PKD management, particularly for reducing active cyst growth. It should, however, be accompanied by active microbiome support, fungal monitoring, and bifidobacterial replenishment, rather than used as a sole intervention.
- Cell Danger Response support: Interventions that support CDR resolution, including mitochondrial support (CoQ10, NAD+ precursors), antioxidant strategies, vagal tone activation, and nervous system regulation, may help shift cells out of chronic defensive metabolic states.
- FMT awareness: While FMT for PKD remains investigational, practitioners should be aware of the emerging evidence base and the forthcoming research from the Friedman Laboratory and associated centres.
Conclusion: A More Intelligent Lens on PKD
PKD has long been framed as an inevitable genetic destiny, a progressive condition with no reversible pathophysiology and limited therapeutic options beyond managing its downstream consequences. The evidence reviewed in this article comprehensively challenges that narrative.
The convergence of the Warburg metabolic signature in both PKD cysts and cancer cells, the endotoxin-as-cyst-trigger evidence, the polycystin-tight junction-leaky tubule mechanism, the CDR framework, the candida-cancer-PKD connection, and the striking microbiome prevalence data together suggest a coherent and testable model: PKD cysts form as a purposeful biological containment response to endotoxin and microbial burden entering renal interstitial tissue through polycystin-haploinsufficient, permeable tubular walls, driven by a gut microbiome that produces the initiating trigger. Address the endotoxin load. Restore the microbiome. Clear the fungal burden. Support the CDR’s resolution. That is where the meaningful therapeutic leverage exists.
The finding that 96% of germ-free mice carrying the PKD gene mutation develop no cysts is not a biological curiosity. It is the central clue. The gut microbiome is not a bystander in PKD. It may be the conductor.
For patients and practitioners working with this condition, this framework offers both a more complete explanation of disease biology and, critically, a more actionable therapeutic roadmap: one oriented toward resolving the underlying drivers of cyst formation rather than managing their consequences.
References & Further Reading
Seyfried, T.N. (2012). Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer. Wiley.
Microbiome of infected cysts, faeces and saliva in ADPKD patients (PMC)
Trigger Warning: Modern Diet, Lifestyle and Environment in ADPKD Progression — MDPI Nutrients
Role of Interstitial Inflammation in the Pathogenesis of Polycystic Kidney Disease — Nephrology
Mechanisms of Cyst Development in Polycystic Kidney Disease — PMC
Structure and Function of Polycystins: Insights into PKD — Nature Reviews Nephrology
Naviaux, R.K. Metabolic Features of the Cell Danger Response — PubMed
Ketogenic Diets Alter the Gut Microbiome, Resulting in Decreased Intestinal Th17 Cells — Cell
Warburg Effect: A Key Metabolic Hallmark of Cancer Cells
Lintern, M. Cancer Resolution
The author would like to thank Libby Shaw and Felix Mueller for their knowledge and help in creating this article.